全氟化合物(PFASs)作为不遵循传统有机污染物“规则”的一类新污染物,仅是微量存在也会对自然生态和人类健康构成潜在威胁。由于C-F键极高的键能,PFASs的化学结构非常稳定,相比于一般卤代有机物,PFASs毒性更高、可生化性更差,很难通过自然分解和微生物代谢被去除。如何长效去除水中多类全氟化合物已成为控制新污染物的新兴热点与难题。物理吸附是目前清除水体中PFASs的主要方法,但后续仍需要处置浓缩后的污染物,无法从环境中彻底消除PFASs。通过破坏部分或全部C-F键降低生态毒性和提高可生化性,是全氟化物污染风险防控更彻底且更可持续的解决方案。目前已有物化催化技术已被证明可以实现全氟化物的有效脱氟,但往往伴随着高成本和严苛的反应条件,且易生成毒性不明或可降解性进一步降低的未知中间产物,进而限制了规模化实际应用。
南京大学环境与健康研究院周琛副教授近年研究证明了基于纳米贵金属的加氢催化过程可在常温常压的温和条件下实现全氟辛酸通过还原脱氟转化为毒性较小的多氟或无氟辛酸,这一突破为全氟化物绿色可控分解转化提供了可行的理论支撑。为突破温和条件下催化脱氟效能难以控制的难题,该研究利用界面精细结构表征结合密度泛函理论计算等手段,建立了常温常压下固(催化剂)— 气(氢能)— 液(污染物)多相界面构效关系,在纳米钯催化以PFOA为代表的全氟长链羧酸脱氟过程中,通过建立单个吸附位点与底物活化方式的对应关系,发现在未形成活化氢的位点,底物羧基端强烈倾向于垂直吸附到钯-氢界面,在空间和能垒上阻断碳-氟键与钯-氢键的接触活化,解释了全氟长链羧酸无法加氢脱氟的关键障碍;而在钯原子表面形成活化氢的位点,底物碳链骨架倾向于平行吸附到钯-氢界面,激活碳-氟键加氢取代反应实现脱氟,进而阐明了固-液界面催化活性位点空间亲密度与固-气界面氢活化程度对催化脱氟活性和选择性的影响规律及作用机理。在基于活性位点分布特征的多相结构基础上,该研究进一步提出了定向调控催化界面活性位点的空间亲密度和活化氢饱和度提高脱氟催化活性和选择性的方法,阐明了固-气界面上氢分子通量可以通过改变纳米催化剂表面氢分子裂解速率影响活化氢饱和度,从而调控梯级加氢脱氟反应的脱氟程度。在催化剂表面活化氢饱和状态下,全氟辛酸及其部分脱氟产物的垂直吸附得到完全抑制,实现完全脱氟为辛酸。该研究为解决双碳背景下全氟类新污染物风险控制提供了新的思路。
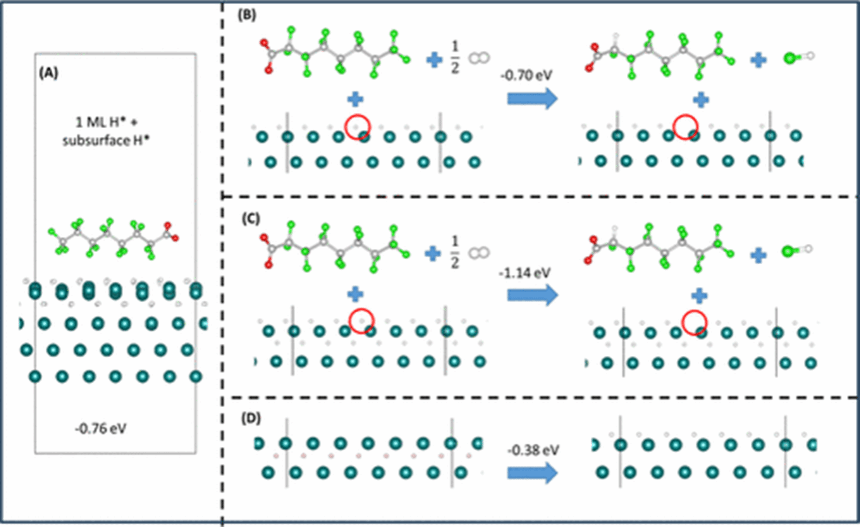
相关研究成果发表在环境类权威期刊Environ. Sci. Technol.上(原文链接:https://pubs.acs.org/doi/10.1021/acs.est.3c07650)。南京大学环境与健康研究院周琛副教授为唯一通讯作者。该研究得到了南京大学高层次人才启动基金等项目支持。





 当前位置:
当前位置:

